The Gut Trimethylamine Cycle: Microbial Ecology, Quantitative Metabolic Flux, and Therapeutic Targeting
published: 18 November 2025 | https://doi.org/10.63174/xdi.HKRH5552
Abstract
Trimethylamine (TMA) and its hepatic oxidation product, trimethylamine N-oxide (TMAO), form a pivotal gut–liver metabolic axis linking the intestinal microbiota with host cardiometabolic health. Elevated plasma TMAO levels have been consistently associated with atherosclerosis, chronic kidney disease, diabetes, and other metabolic disorders. This review systematically summarizes current advances in understanding the microbial ecology and biochemical pathways underlying the gut TMA cycle. We highlight three major microbial routes for TMA generation—cutC/D-mediated choline cleavage, cntA/B-dependent carnitine oxidation, and BbuA-associated γ-butyrobetaine reduction - as well as methanogenic and acetogenic pathways responsible for TMA utilization and detoxification. Integrating multi-omics data with metabolic-flux modeling provides quantitative insight into the gut–liver TMA network, revealing key rate-limiting nodes and interindividual variability driven by diet and microbial composition. Emerging intervention strategies include chemical inhibition of cutC/D, cntA/B activity, dietary modulation, probiotic or engineered consortia enhancing TMA clearance, and bacteriophage-based precision targeting of TMA-producing taxa. Finally, we propose developing a gut TMA index and mapping producer–degrader interaction networks as frameworks for personalized risk evaluation and therapeutic design. Collectively, quantitative and translational studies of the TMA cycle are expected to establish new paradigms for microbiome-driven prevention and treatment of cardiometabolic diseases.
1. Introduction
Trimethylamine (TMA) and its oxidative product, trimethylamine N-oxide (TMAO), have emerged over the past decade as critical metabolic signals linking the gut microbiota with host metabolic health[1]. Numerous clinical and experimental studies have demonstrated that elevated plasma TMAO levels are closely associated with the development of atherosclerosis, chronic kidney disease, diabetes, and neurodegenerative disorders[2,3]. Because TMAO originates exclusively from microbial metabolism of dietary methylamine-containing compounds such as choline, carnitine, and betaine, the gut microbiota plays a decisive role in TMA production and regulation[4].
Within the gut–liver axis, specific anaerobes first cleave dietary precursors to release TMA; the metabolite then enters the liver via the portal vein, where it is oxidized by flavin-containing monooxygenase 3 (FMO3) to generate TMAO before entering systemic circulation[5]. This process forms a prototypical gut–liver metabolic interaction loop, illustrating how microbial activity directly shapes host cardiovascular and metabolic homeostasis. However, the composition, functional diversity, and dynamic balance of TMA-producing and TMA-degrading communities in the gut remain insufficiently characterized. Current research has largely focused on TMA biosynthesis, while the microbial pathways responsible for its degradation and reutilization remain poorly understood[6]. Notably, several facultative or opportunistic pathogens—such as Klebsiella pneumoniae and Clostridium sporogenes can generate large amounts of TMA via the cutC/D, cntA/B pathways, potentially contributing to TMAO-associated pathogenesis[7]. Conversely, certain methanogenic archaea and acetogenic bacteria can utilize TMA as a methyl donor, converting it into methane or acetate, thereby reducing luminal TMA load and maintaining metabolic equilibrium[8]. Identifying and quantifying these “TMA-producing” and “TMA-consuming” microorganisms is therefore crucial for understanding the ecological regulation of the gut TMA network.
This review aims to provide a comprehensive overview of the microbial foundations of the gut TMA cycle, emphasizing the pathways and key taxa involved in TMA production and degradation. It further introduces a quantitative flux-based framework for studying TMA metabolism and discusses the potential of TMA-associated microbial populations as targets for therapeutic modulation of TMAO-related diseases.
2. Microbial Mechanisms and Ecological Regulation of Gut TMA Formation
Collectively, microbial transformation of dietary methylamine substrates forms the gut–liver TMA metabolic axis, in which choline and carnitine are cleaved by the cutC/D or cntA/B systems to produce TMA, subsequently oxidized by hepatic FMO3 into TMAO (Figure 1).
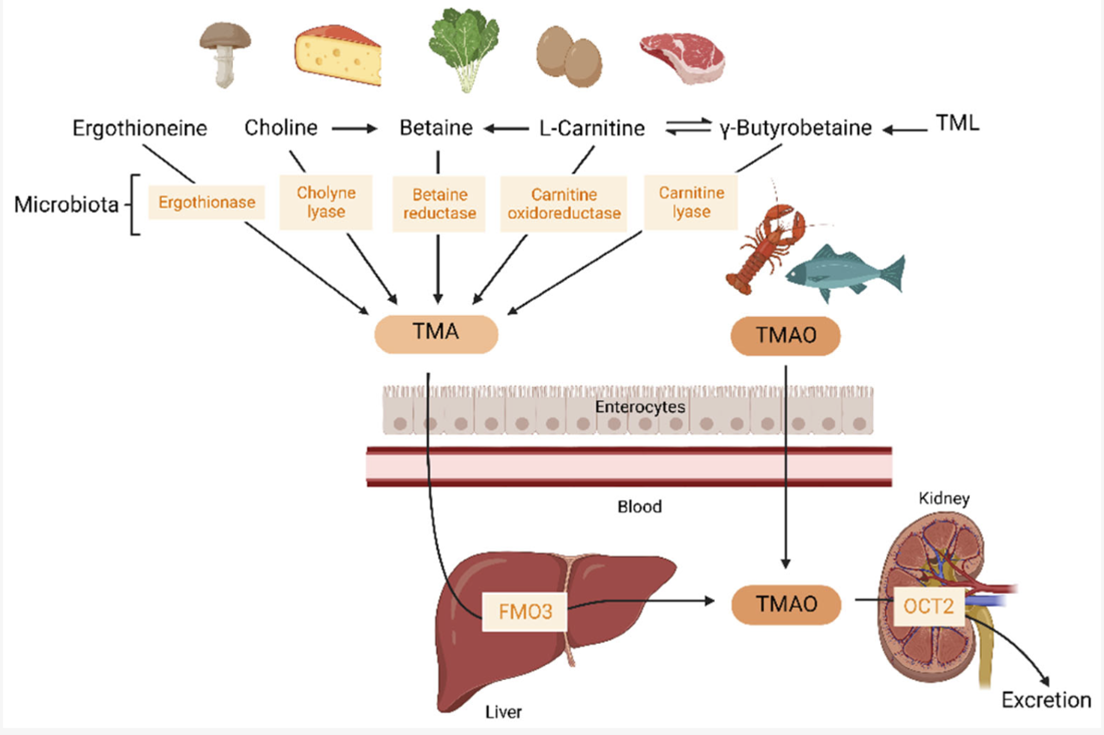
Figure 1. Biochemical overview of the gut–liver TMA–TMAO metabolic pathway. Dietary precursors such as choline and carnitine are converted by gut microbial enzymes (CutC/CutD or CntA/CntB systems) into trimethylamine (TMA), which diffuses into the portal circulation and is oxidized by hepatic flavin-containing monooxygenase 3 (FMO3) to form trimethylamine-N-oxide (TMAO). Circulating TMAO contributes to cardiometabolic and renal disease risk by modulating vascular, inflammatory, and lipid pathways. Figure adapted from Wang et al., Int. J. Mol. Sci. 2023, 24(19), 1940 (CC BY 4.0).
2.1. Choline Metabolism Pathway (cutC/cutD System)
Choline is the earliest identified precursor for microbial TMA formation. This reaction is primarily mediated by anaerobic bacteria such as Clostridium sporogenes, Desulfovibrio desulfuricans, and Klebsiella pneumoniae. The key enzyme cutC is a [4Fe–4S] cluster-containing glycyl radical enzyme that, upon activation by cutC/D, catalyzes the C–N bond cleavage of choline to yield TMA and acetaldehyde[9]. The reaction is highly oxygen-sensitive and occurs efficiently only under low-oxygen or strictly anaerobic conditions. Metagenomic surveys show that the cutC/D gene is widely distributed, with detection rates of approximately 2–8% across human cohorts and relative abundances ranging from 10⁻⁶ to 10⁻⁵[10]. Its abundance and transcription are elevated in individuals consuming choline- or fat-rich diets and positively correlate with plasma TMAO levels[11]. These findings indicate that the cutC/D pathway constitutes a major contributor to total TMA output and may play a pro-atherogenic role in cardiometabolic diseases.
2.2. Carnitine Oxidation Pathway (cntA/cntB System)
Carnitine can be oxidatively deaminated by the cntA/B enzyme complex in certain facultative anaerobes, producing TMA and malic semialdehyde[12]. This reaction requires FAD and molecular oxygen as cofactors, making it most active in the microaerobic regions of the intestinal mucosa. Representative strains include Acinetobacter calcoaceticus and Desulfovibrio alaskensis, which typically colonize mucosal surfaces or oxygen-gradient interfaces, showing increased activity under high-protein or meat-rich diets[13]. Population-level analyses reveal cntA/B detection rates of 1.9–6.7% and relative abundances between (1.6 × 10⁻⁶ – 2.6 × 10⁻⁵), positively correlated with cutC abundance. This suggests potential ecological complementarity between the two pathways, collectively determining TMA production capacity[14].
2.3. γ-Butyrobetaine Reduction Pathway (BbuA/BbuB/BbuC System)
γ-Butyrobetaine, an intermediate of carnitine metabolism, can be reductively cleaved to TMA and butyrate by Eubacterium species and Emergencia timonensis through the bbuA/B/C gene cluster[15]. This pathway operates under strictly anaerobic conditions and is coupled to short-chain fatty acid (SCFA) formation, implying a potential link to host energy metabolism. Current metagenomic data remain limited, but preliminary evidence indicates that bbuA is detected at much lower frequency than cutC/D or cntA/B, likely enriched only within specific Firmicutes subgroups[12].
2.4. Environmental and Ecological Determinants
Gut TMA formation exhibits marked functional redundancy and ecological dependence. Although cutC/D and cntA/B are broadly distributed across taxa, their transcription and enzymatic activity are strongly modulated by environmental factors. Parameters such as pH, substrate competition, sulfate, and nitrogen availability all influence metabolic preference. For instance, when sulfate is abundant, Desulfovibrio preferentially performs sulfate respiration over choline cleavage, thereby reducing TMA yield[16]. Variations in hydrogen (H₂) levels also modulate flux direction and rate: H₂ accumulation maintains a reductive environment favoring the cutC/D pathway, whereas more oxidizing conditions enhance cntA/B activity[9]. This dynamic regulation underlies the pronounced interindividual and temporal variability of intestinal TMA production.
2.5. Pathway Contribution and Quantitative Relevance
The relative contribution of each substrate pathway to total TMA output varies with diet, microbiome composition, and metabolic milieu. Based on metabolic flux modeling and stable isotope tracing, the choline pathway contributes roughly 30–40%, while the carnitine and γ-butyrobetaine pathways together account for approximately 50–60% of total TMA production; the remainder likely derives from minor or uncharacterized sources[17]. Establishing quantitative models linking cutC/D, cntA/B, and bbuA/B/C gene abundances with measured TMA levels is thus essential for elucidating TMA flux dynamics and individual disease risk.
2.6. Intestinal Metabolism and Clearance of TMA
Not all TMA produced in the gut enters the portal circulation. A portion is further metabolized or assimilated by other microbes, reducing hepatic load. The main route involves methanogenic archaea such as Methanomassiliicoccus luminyensis and Methanosarcina barkeri, which use TMA methyl groups via the mttB/mcr pathway to generate methane while releasing ammonia[18]. Additionally, acetogenic bacteria like Acetobacterium woodii can demethylate TMA stepwise to dimethylamine (DMA) and monomethylamine (MMA), assimilating nitrogen into amino acids or converting it to acetate through the Wood - Ljungdahl pathway[19]. These reactions become more prominent under hydrogen-limited or mildly oxidizing conditions and act as microbial “sinks” that sequester TMA. Consequently, the actual intestinal TMA flux depends on the dynamic balance between producers and degraders - the greater the activity of the latter, the lower the fraction of TMA entering systemic circulation.
3. Representative TMA-Producing Bacteria and Population Features
Gut bacteria carrying key genes such as cutC/D and cntA/B constitute the principal ecological units responsible for TMA generation. Metagenomic surveys reveal that these microorganisms are distributed across multiple phyla, including Proteobacteria, Firmicutes, and Actinobacteria, and display substantial interindividual diversity within human populations[20].
Choline-degrading communities are represented by Clostridium sporogenes, Desulfovibrio desulfuricans, and Klebsiella pneumoniae, which are commonly found in the anaerobic regions of the colon. Through the cutC/D system, these species cleave choline to produce TMA, showing increased abundance in individuals with high-choline diets, inflammatory bowel disease, or metabolic disorders[20]. Some studies report enhanced activity of these bacteria in mucosal or hemolytic microenvironments, suggesting that they may gain ecological advantages under local oxidative stress.
Carnitine-oxidizing bacteria mainly include members of the Acinetobacter and Desulfovibrio genera harboring the cntA/B system, which operate within the microaerobic mucosal layer to convert carnitine into TMA[21]. Their abundance positively correlates with plasma TMAO levels in hosts consuming red meat–rich diets or experiencing oxidative stress[22]. Additional taxa, such as Enterobacter cloacae and Proteus mirabilis, also possess potential TMA-producing capacity, although their contribution depends on dietary composition and the gut redox environment.
Population-level data indicate that TMA-producing bacteria generally remain at low abundance in healthy individuals but increase markedly in patients with metabolic syndrome, renal impairment, or cardiovascular disease[23]. This enrichment likely reflects both dietary and metabolic influences, as well as a possible role of these taxa in disease progression. Nevertheless, they cannot be classified simplistically as “pathogenic,” since under normal ecological conditions they also contribute to nitrogen turnover and short-chain fatty acid production. Overall, bacterial populations carrying the cutC/cntA systems exhibit notable metabolic plasticity across ecological niches, and their abundance dynamics may serve as useful indicators of gut TMA cycle activity.
4. Representative TMA-Utilizing and Transforming Microbes
In contrast to TMA-producing populations, certain gut microorganisms can metabolize TMA through reductive or demethylation reactions, thereby limiting its accumulation within the intestinal lumen. These taxa mainly include methanogenic archaea and acetogenic bacteria, whose distribution and activity are influenced by hydrogen availability, redox potential, and substrate competition.
Methanogenic archaea, such as Methanomassiliicoccus luminyensis and Methanosarcina barkeri, utilize the mttB/mcr enzymatic system to transfer the methyl group of TMA onto coenzyme M, ultimately producing methane and ammonia[24]. This process depends on hydrogen-rich, low-redox conditions and forms a syntrophic relationship with hydrogen-producing fermenters. Metagenomic analyses indicate that the mttB gene is detectable in approximately 15–25 % of human samples, often showing an inverse correlation with cutC/cntA abundance[25]. Such reciprocity suggests potential metabolic competition or ecological balancing between TMA producers and consumers.
Acetogenic bacteria, including Acetobacterium woodii and Eubacterium limosum, demethylate TMA stepwise via mta/mtt-type methyltransferases, converting it through the Wood–Ljungdahl pathway into acetate[26],[27]. This process both removes TMA and generates short-chain fatty acids usable by the host, underscoring its importance in coupled carbon–nitrogen cycling. Although typically of low abundance, these acetogens exhibit markedly enhanced activity under anaerobic, hydrogen-rich gut conditions. Collectively, TMA-utilizing and transforming microbes act as metabolic buffers that maintain intestinal homeostasis. By channeling TMA into methane or acetate, they effectively reduce its efflux to the liver and modulate systemic TMAO exposure.
5. Quantitative Insights into the Gut TMA Metabolic Network
The gut TMA metabolic network represents a cross-species and multi-layer co-metabolic system whose dynamic balance determines systemic exposure to TMAO—a metabolite repeatedly linked to the risks of atherosclerosis, chronic kidney disease, and metabolic syndrome[28],[29]. However, most clinical research remains focused on the correlation between plasma TMAO levels and disease phenotypes, without elucidating the underlying microbe–host metabolic kinetics. Achieving predictive and actionable translational applications requires a quantitative understanding of TMA metabolic flux. Quantitative flux analysis can characterize the rate balance between microbial TMA production and host oxidation, identify “high-flux nodes” and “bottleneck reactions,” and provide quantitative support for dietary modulation, microbiome intervention, and drug target discovery[10],[30]. Thus, flux quantification of the TMA cycle has become a critical bridge linking mechanistic microbiome research with clinical risk assessment and serves as the theoretical foundation for metabolic intervention strategies.
5.1. Research Needs: Multi-Layer Integration of TMA Flux
Quantitative studies of TMA metabolism aim to determine the reaction rates and relative contributions of each step—from substrate input and microbial cleavage to host oxidation. This requires integration of multi-omics data, including metagenomics (microbial gene potential), metatranscriptomics (gene expression activity), and metabolomics (TMA/TMAO concentrations), to construct a system-level analytical framework. Such integrative approaches can reveal: (i) interindividual differences in the functional strength of cutC/D, cntA/B, bbuA/B/C modules; (ii) dynamic coupling between intestinal and plasma TMAO concentrations; and (iii) the impact of specific dietary or pharmacological interventions on flux balance[32].
5.2. Methodological Strategies: Integrating Multi-Omics and Modeling
The workflow for TMA flux quantification typically involves three key steps:Gene-level inference: Estimating the relative abundance and transcriptional activity of key enzymatic genes (cutC/D, cntA/B, bbuA/B/C) from metagenomic or metatranscriptomic data to infer microbial TMA-producing potential[10],[12]. Metabolite quantification: Measuring TMA, TMAO, and intermediate compounds (e.g., DMA, MMA) in fecal and plasma samples using LC–MS/MS, complemented by isotope-tracing experiments to determine in vivo and in vitro conversion rates[33]. Flux modeling: Constructing an integrated gut–liver TMA metabolic network using flux balance analysis (FBA) or Gibbs energy–constrained models to couple microbial metabolism with hepatic oxidation[34]. This framework enables quantitative estimation of pathway-specific fluxes (e.g., cutC/D-mediated choline cleavage) and prediction of steady-state distributions under varying ecological and host conditions.
5.3. Key Challenges and Future Directions
Current challenges include:
(1) Functional redundancy – Many species harbor similar TMA-producing gene clusters, making single-gene abundance a poor proxy for actual metabolic capacity[12].
(2) Environmental dependency – TMA production and demethylation reactions are highly sensitive to redox potential, substrate concentration, and hydrogen partial pressure, leading to sample-specific variability in flux estimation[35].
(3) Cross-scale integration – Gut microbial metabolism, hepatic oxidation, and systemic circulation occur on distinct spatial and temporal scales, requiring standardized parameters and validation systems for accurate model coupling[36].
Future progress may come from integrating ex vivo gut ecosystem models with steady-state isotope flux tracing (¹³C/¹⁵N), which would enhance both the accuracy and interpretability of TMA flux modeling.
6. Targeting the TMA Cycle: Therapeutic Perspectives
Gut TMA metabolism represents not only a microbe–host co-metabolic process but also a key intersection in the risk landscape of chronic diseases. Elevated plasma TMAO levels have been firmly linked to the development of atherosclerosis, chronic kidney disease, diabetes, and heart failure. Clinical studies further indicate that TMAO remains an independent predictor of cardiometabolic disease risk even after adjusting for diet, age, and BMI[37]. Therefore, strategies that target the TMA cycle are increasingly viewed as a bridge between microbiome research and metabolic disease prevention. Compared with traditional host-centric approaches, microecological targeting offers advantages of reversibility, tunability, and minimal systemic side effects, making it a promising direction for precision intervention (Figure 2).
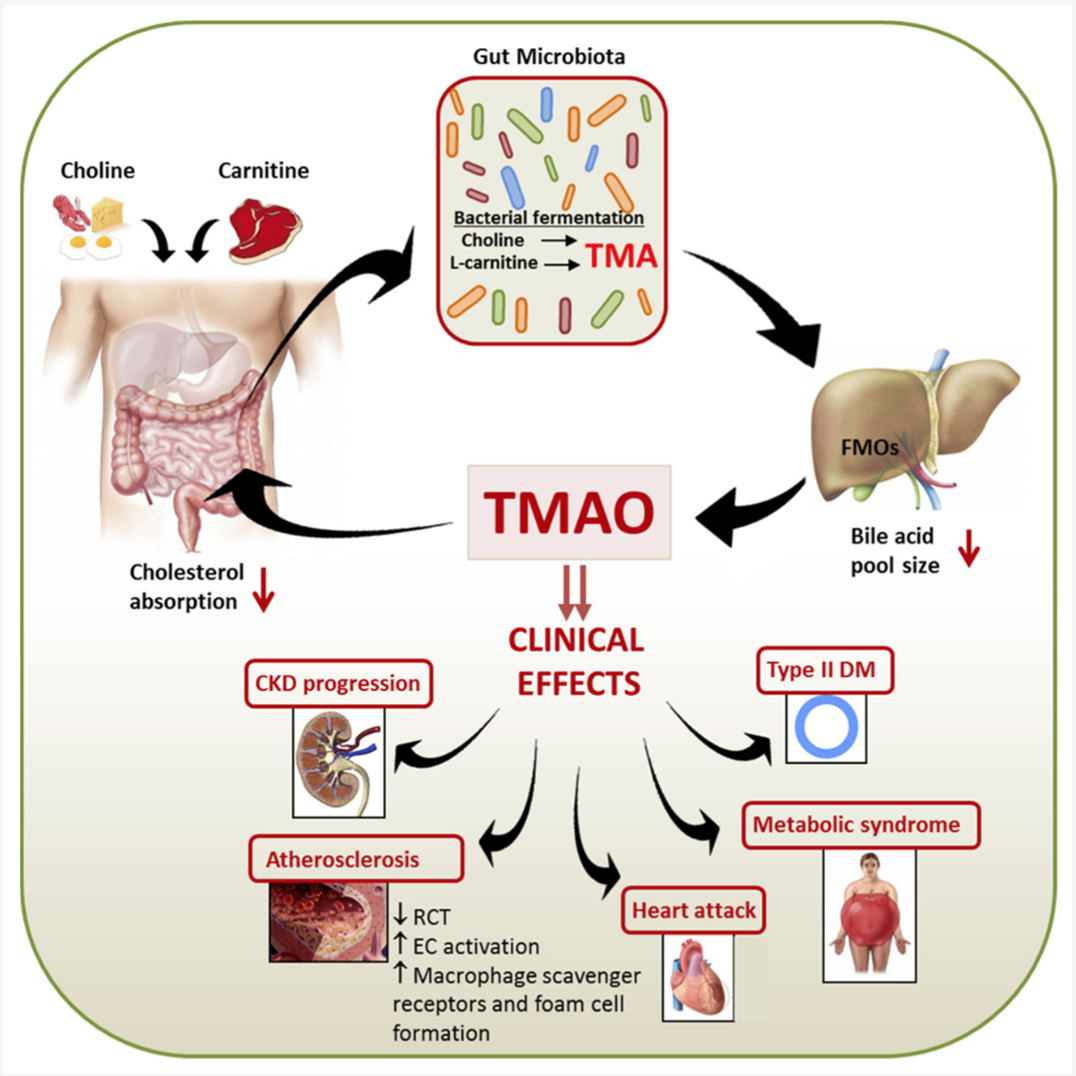
Figure 2. The gut–liver trimethylamine (TMA)–trimethylamine N-oxide (TMAO) axis and its pathological relevance. Dietary precursors such as choline, carnitine, and betaine are metabolized by gut microbiota to generate TMA, which enters the portal circulation and is oxidized by hepatic FMO3 into TMAO. Circulating TMAO contributes to endothelial dysfunction, lipid accumulation, platelet activation, and chronic inflammation, thereby promoting cardiovascular, renal, and metabolic diseases. Elevated plasma TMAO also serves as a diagnostic and prognostic biomarker for cardiometabolic risk. Figure adapted from Cho et al., Toxins 2016, 8(10): 326 (CC BY 4.0).
6.1. Inhibitory Strategies: Blocking Upstream TMA Formation
Direct inhibition of intestinal TMA formation is the earliest and most straightforward approach. The representative compound 3,3-dimethyl-1-butanol (DMB) acts as a choline analog that competitively binds the cutC/D enzyme, markedly reducing choline cleavage rates[38]. Animal studies have shown that DMB treatment lowers plasma TMAO levels by 50–70% and mitigates atherosclerotic plaque formation[39]. Beyond DMB, small-molecule screening has identified additional inhibitors targeting cutC/D or cntA/B enzymes, including halogenated choline derivatives and FAD-competitive compounds[40]. In addition, dietary modification remains the most practical and accessible intervention. Reducing the intake of choline- and carnitine-rich foods—such as red meat, egg yolk, and full-fat dairy products—can effectively lower substrate availability and thereby decrease upstream TMA production[41]. Although long-term adherence and metabolic compensation require further evaluation, the safety and general applicability of dietary regulation make it a robust foundation for clinical exploration.
6.2. Enhancement Strategies: Promoting Downstream TMA Clearance
An alternative strategy is to enhance microbial TMA consumption or transformation. First, TMA-degrading methanogens such as Methanomassiliicoccus luminyensis have been shown to utilize TMA in vitro, converting it to methane and ammonia, suggesting potential as probiotic candidates[42]. Reconstruction or enrichment of these communities could intercept TMA flux before it enters the circulation. Second, advances in synthetic biology and microbial community engineering enable the design of synthetic consortia in which TMA producers and consumers are co-cultured to achieve stable metabolic equilibria, effectively “flattening” TMA peaks under simulated gut conditions[43]. Furthermore, metabolic engineering of nonpathogenic chassis strains to express mttB/mcr or related TMA-utilization pathways can endow them with competitive uptake and catabolic capacity for TMA[44]. This approach has been shown in animal models to reduce plasma TMAO levels and improve lipid metabolism, though its long-term ecological safety requires further validation.
6.3. Phage Therapy Prospects: Targeting Specific TMA-Producing Bacteria
Recently, bacteriophage therapy has emerged as a precision tool for species-specific modulation of gut microbial metabolism. Unlike broad-spectrum antibiotics, phages can selectively eliminate TMA-producing bacteria such as Klebsiella pneumoniae and Desulfovibrio desulfuricans without disrupting the overall microbial ecosystem[45]. Animal experiments demonstrate that administration of K. pneumoniae-targeting phages significantly lowers intestinal TMA levels and alleviates diet-induced atherosclerosis[46]. In the future, engineered phages incorporating CRISPR–Cas delivery systems may enable functional suppression of cutC/D, cntA/B genes, effectively “silencing” TMA-producing activity without eradicating host bacteria[47]. Such strategies balance specificity with ecological stability and are considered among the most promising translational avenues for treating microbiome-related metabolic diseases.
6.4. Translational Perspectives and Future Clinical Applications
Advancing research on the TMA metabolic cycle toward clinical translation requires an integrated framework that brings together evidence from prospective cohorts, quantitative metabolic assessment, and precision microbiome interventions. First, large-scale longitudinal studies remain essential to clarify the disease spectrum associated with dysregulated TMA/TMAO metabolism and to establish its independent predictive value beyond diet, renal function, and conventional cardiovascular risk factors. Such cohorts will help identify individuals who exhibit persistently elevated TMA production or impaired clearance and who may therefore benefit from targeted modulation of the TMA pathway.
Second, static plasma TMAO concentrations alone are insufficient to capture the underlying metabolic dynamics. A shift toward flux-based evaluation is needed. A composite TMA balance index—incorporating microbial TMA-producing capacity (cutC/D, cntA/B, bbuA/B/C), microbial TMA-degrading pathways (e.g., mttB/mcr systems), and host oxidative and renal handling—may provide a more robust and clinically actionable measure of TMA metabolic disturbance. Such an index could support individualized risk stratification, therapeutic monitoring, and prognostic assessment in cardiometabolic settings.
In addition, precision microbiome modulation has emerged as a promising therapeutic direction. Drawing on established workflows for phage therapy against multidrug-resistant pathogens, phage-based targeting of TMA-producing bacteria could follow a similar translational pathway: microbiome-guided patient identification, phage isolation and host-range testing, ecological assessment in synthetic gut systems, and early-phase clinical evaluation of safety and metabolic impact. Looking ahead, engineered phages capable of selectively suppressing cutC/cntA activity may further reduce TMA production while preserving broader microbial community stability.
Collectively, these prospective research and intervention strategies outline a coherent route toward clinical translation, offering new opportunities for prediction, monitoring, and targeted modulation of TMA-associated cardiometabolic risk.
7. Conclusion and Future Directions
The gut TMA cycle represents a critical interface between host metabolism, the microbiota, and dietary inputs. As a prototypical co-metabolic system, it not only determines circulating TMAO levels but also plays a central role in the pathogenesis of cardiovascular, renal, and metabolic diseases. Recent genomic and metabolomic studies of TMA-producing and TMA-degrading microbial populations have begun to uncover the multilayered regulatory mechanisms of this system, providing a quantitative foundation for metabolic intervention. Future research should focus on quantification and systems integration. First, multi-omics approaches could be used to establish a gut TMA index that reflects both community composition and metabolic flux intensity, enabling individualized assessment of TMA turnover and disease risk. Second, constructing a TMA producer–degrader interaction network will clarify the ecological interactions and competition among functional guilds. Finally, advancing the clinical translation of TMA-targeted interventions including metabolic modulation, microbiome reconstruction, and phage-based therapies will be crucial for moving from descriptive correlations to actionable mechanisms. Moreover, quantitative flux modeling may help predict individual TMA responses to specific dietary patterns, thereby informing personalized nutritional strategies. Likewise, integrating flux-based predictions with microbial-targeted approaches, such as phage-based modulation or synthetic microbiome reconstruction may help estimate the achievable reduction in TMA generation following precision ecological interventions. In summary, quantitative and interventional studies of the TMA cycle will provide a new paradigm for understanding host–microbiome metabolic interactions and open new avenues for precision prevention and treatment of metabolism-related diseases.
Author Contributions
Xin Xing contributed to conceptualization, literature review, data interpretation, and manuscript drafting. Yangyang Zhao supervised the study, provided critical revisions, and finalized the manuscript. Both authors read and approved the final version of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Funding
No funding.
Acknowledgements
The authors gratefully acknowledge the collective contributions of all co-authors to the conception, discussion, and refinement of this review. Each author's expertise and collaboration were essential to the completion of this work.
References
-
Y.H. Zhou, Y.W. Zhang, S.K. Jin, J. Lv, M.L. Li, N.H. Feng. "The gut microbiota derived metabolite trimethylamine N-oxide: Its important role in cancer and other diseases." Biomed. Pharmacother. 2024, 8, 177, 117031.
-
X.S. Meng, Z. Li, M. Wang, M.C. de Oliveira Otto, R.N. Lemaitre, A. Fretts, N. Sotoodehnia, M. Budoff, I. Nemet, J.A. DiDonato, W.H.W. Tang, B.M. Psaty, D.S. Siscovick, S.L. Hazen, D. Mozaffarian. "Trimethylamine N-oxide is associated with long-term mortality risk: the multi-ethnic study of atherosclerosis." Eur. Heart J. 2023, 18, 44, 1608-1618.
-
K. Jaworska, M. Kuś, M. Ufnal. "TMAO and diabetes: from the gut feeling to the heart of the problem." Nutr. Diabetes 2025, 1, 15, 21.
-
J. Liu, P. Ge, Y. Luo, Z. Sun, X. Luo, H. Li, B. Pei, L. Xun, X. Zhang, Y. Jiang, H. Wen, J. Liu, Q. Yang, S. Ma, H. Chen. "Decoding TMAO in the gut-organ axis: from biomarkers and cell death mechanisms to therapeutic horizons." Drug Des. Devel. Ther. 2025, 29, 19, 3363-3393.
-
J.W. Jang, E. Capaldi, T. Smith, P. Verma, J. Varga, K.J. Ho. "Trimethylamine N-oxide: a meta-organismal axis linking the gut and fibrosis." Mol. Med. 2024, 1, 30, 128.
-
Y.R. Chen, L.D. Chen, L.J. Zheng. "Exploring the trimethylamine-degrading genes in the human gut microbiome." AMB Express 2025, 1, 15, 91.
-
Y. He, S. Chen, Y. Xue, H. Lu, Z. Li, X. Jia, Y. Ning, Q. Yuan, S. Wang. "Analysis of alterations in intestinal flora in Chinese elderly with cardiovascular disease and its association with trimethylamine." Nutrients 2024, 12, 16, 1864.
-
G. Borrel, A. McCann, J. Deane, M.C. Neto, D.B. Lynch, J.-F. Brugère, P.W. O'Toole. "Genomics and metagenomics of trimethylamine-utilizing Archaea in the human gut microbiome." ISME J. 2017, 9, 11, 2059-2074.
-
Y.Q. Yang, W.H. Deng, R.Z. Liao. "Mechanistic insights into choline degradation catalyzed by the choline trimethylamine-lyase CutC." J. Phys. Chem. B 2025, 22, 129, 5438-5448.
-
S. Rath, B. Heidrich, D.H. Pieper, M. Vital. "Uncovering the trimethylamine-producing bacteria of the human gut microbiota." Microbiome 2017, 1, 5, 54.
-
M. Haas, B. Brandl, K. Neuhaus, S. Wudy, K. Kleigrewe, H. Hauner, T. Skurk. "Effect of dietary fiber on trimethylamine-N-oxide production after beef consumption and on gut microbiota: MEATMARK - a randomized cross-over study." Eur. J. Clin. Nutr. 2025, 10, 79, 980-990.
-
S. Rath, T. Rud, D.H. Pieper, M. Vital. "Potential TMA-producing bacteria are ubiquitously found in Mammalia." Front. Microbiol. 2019, 10, 2966.
-
M. Castanheira, R.E. Mendes, A.C. Gales. "Global epidemiology and mechanisms of resistance of Acinetobacter baumannii-calcoaceticus complex." Clin. Infect. Dis. 2023, Suppl 2, 76, S166-S178.
-
Y.Y. Cai, F.Q. Huang, X. Lao, Y. Lu, X. Gao, R.N. Alolga, K. Yin, X. Zhou, Y. Wang, B. Liu, J. Shang, L.-W. Qi, J. Li. "Integrated metagenomics identifies a crucial role for trimethylamine-producing Lachnoclostridium in promoting atherosclerosis." NPJ Biofilms Microbiomes 2022, 1, 8, 11.
-
L.J. Rajakovich, B. Fu, M. Bollenbach, E.P. Balskus. "Elucidation of an anaerobic pathway for metabolism of L-carnitine-derived γ-butyrobetaine to trimethylamine in human gut bacteria." Proc. Natl. Acad. Sci. U.S.A. 2021, 32, 118, e2101498118.
-
S. Craciun, E.P. Balskus. "Microbial conversion of choline to trimethylamine requires a glycyl radical enzyme." Proc. Natl. Acad. Sci. U.S.A. 2012, 52, 109, 21307-21312.
-
P. DayWalsh, E. Shehata, S. Saha, G.M. Savva, B. Nemeckova, J. Speranza, L. Kellingray, A. Narbad, P.A. Kroon. "The use of an in-vitro batch fermentation (human colon) model for investigating mechanisms of TMA production from choline, L-carnitine and related precursors by the human gut microbiota." Eur. J. Nutr. 2021, 7, 60, 3987-3999.
-
A. Ramezani, T.D. Nolin, I.R. Barrows, M.G. Serrano, G.A. Buck, R. Regunathan-Shenk, R.E. West, P.S. Latham, R. Amdur, D.S. Raj. "Gut colonization with methanogenic Archaea lowers plasma trimethylamine N-oxide concentrations in apolipoprotein e−/− mice." Sci. Rep. 2018, 1, 8, 14752.
-
R. Jiang, D.J. Kountz, L. Zhang, J.A. Krzycki. "A cobalamin-dependent pathway of choline demethylation from the human gut acetogen Eubacterium limosum." J. Biol. Chem. 2025, 6, 301, 108524.
-
L. Hoyles, M.L. Jiménez-Pranteda, J. Chilloux, F. Brial, A. Myridakis, T. Aranias, C. Magnan, G.R. Gibson, J.D. Sanderson, J.K. Nicholson, D. Gauguier, A.L. McCartney, M.-E. Dumas. "Metabolic retroconversion of trimethylamine N-oxide and the gut microbiota." Microbiome 2018, 1, 6, 73.
-
F. Piskol, P. Lukat, L. Kaufhold, A. Heger, W. Blankenfeldt, D. Jahn, J. Moser. "Biochemical and structural elucidation of the L-carnitine degradation pathway of the human pathogen Acinetobacter baumannii." Front. Microbiol. 2024, 15, 1446595.
-
W.K. Wu, Y.L. Lo, J.Y. Chiu, C.L. Hsu, I.-H. Lo, S. Panyod, Y.C. Liao, T.H.T. Chiu, Y.T. Yang, H.C. Kuo, H.B. Zou, Y.H. Chen, H.L. Chuang, J.J.Y. Yen, J.T. Wang, H.M. Chiu, C.C. Hsu, C.H. Kuo, L.Y. Sheen, H.L. Kao, M.-S. Wu. "Gut microbes with the gbu genes determine TMAO production from L-carnitine intake and serve as a biomarker for precision nutrition." Gut Microbes 2025, 1, 17, 2446374.
-
G. Falony, S. Vieira-Silva, J. Raes. "Microbiology meets big data: the case of gut microbiota-derived trimethylamine." Annu. Rev. Microbiol. 2015, 69, 305-321.
-
L. Dziewit, A. Pyzik, K. Romaniuk, A. Sobczak, P. Szczesny, L. Lipinski, D. Bartosik, L. Drewniak. "Novel molecular markers for the detection of methanogens and phylogenetic analyses of methanogenic communities." Front. Microbiol. 2015, 6, 694.
-
M. Guo, L. Fang, M. Chen, J. Shen, Z. Tan, W. He. "Dysfunction of cecal microbiota and CutC activity in mice mediating diarrhea with kidney-yang deficiency syndrome." Front. Microbiol. 2024, 15, 1354823.
-
D. Litty, F. Kremp, V. Müller. "One substrate, many fates: different ways of methanol utilization in the acetogen Acetobacterium woodii." Environ. Microbiol. 2022, 7, 24, 3124-3133.
-
J.B. Ellenbogen, R. Jiang, D.J. Kountz, L. Zhang, J.A. Krzycki. "The MttB superfamily member MtyB from the human gut symbiont Eubacterium limosum is a cobalamin-dependent γ-butyrobetaine methyltransferase." J. Biol. Chem. 2021, 5, 297, 101327.
-
S.C. Schugar, M.A. Shih, D.S. Warrier, E.A. Helsley, M.G. Burrows, K. Ferguson, Z. Zhang, S.A. Smith, S. Wang, B.C. Li, A.K. Brown. "Trimethylamine and trimethylamine N-oxide, a flavin-containing monooxygenase 3 (FMO3)-mediated host-microbiome metabolic axis implicated in health and disease." Drug Metab. Dispos. 2016, 11, 44, 1839-1850.
-
E. Tacconi, G. Palma, D. De Biase, A. Luciano, M. Barbieri, F. de Nigris, F. Bruzzese. "Microbiota effect on trimethylamine N-oxide production: from cancer to fitness - a practical preventing recommendation and therapies." Nutrients 2023, 3, 15, 563.
-
L. Ramireddy, H.Y. Tsen, Y.C. Chiang, C.Y. Hung, F.C. Chen, H.T. Yen. "The gene expression and bioinformatic analysis of choline trimethylamine-lyase (CutC) and its activating enzyme (CutD) for gut microbes and comparison with their TMA production levels." Curr. Res. Microb. Sci. 2021, 2, 100043.
-
M.A. Wörheide, J. Krumsiek, G. Kastenmüller, M. Arnold. "Multi-omics integration in biomedical research - a metabolomics-centric review." Anal. Chim. Acta 2021, 1141, 144-162.
-
Q. Wu, Y. Zhao, X. Zhang, X. Yang. "A faster and simpler UPLC-MS/MS method for the simultaneous determination of trimethylamine N-oxide, trimethylamine and dimethylamine in different types of biological samples." Food Funct. 2019, 10, 6484-6491.
-
A. Sahu, M.-A. Blätke, J.J. Szymański, N. Töpfer. "Advances in flux balance analysis by integrating machine learning and mechanism-based models." Comput. Struct. Biotechnol. J. 2021, 19, 4626-4640.
-
N. Arias, S. Arboleya, J. Allison, A. Kaliszewska, S.G. Higarza, M. Gueimonde, J.L. Arias. "The relationship between choline bioavailability from diet, intestinal microbiota composition, and its modulation of human diseases." Nutrients 2020, 12, 8, 2340.
-
P. Roberts, J. Basran, E.K. Wilson, R. Hille, N.S. Scrutton. "Redox cycles in trimethylamine dehydrogenase and mechanism of substrate inhibition." Biochemistry 1999, 38, 45, 14927-14940.
-
J. Shen, W. Liang, R. Zhao, Y. Chen, Y. Liu, W. Cheng, T. Chai, Y. Zhang, S. Chen, J. Liu, X. Chen, Y. Deng, Z. Zhang, Y. Huang, H. Yang, L. Pang, Q. Qiu, H. Deng, S. Pan, L. Wang, J. Ye, W. Luo, X. Jiang, X. Huang, W. Li, E.L.-H. Leung, L. Zhang, L. Huang, Z. Yang, R. Chen, J. Mei, Z. Yue, H. Wei, K. Kristiansen, L. Han, X. Fang. "Cross-tissue multi-omics analyses reveal the gut microbiota's absence impacts organ morphology, immune homeostasis, bile acid and lipid metabolism." iMeta 2025, 4, 1, e272.
-
L. Guasti, S. Galliazzo, M. Molaro, E. Visconti, B. Pennella, G.V. Gaudio, A. Lupi, A.M. Grandi, A. Squizzato. "TMAO as a biomarker of cardiovascular events: a systematic review and meta-analysis." Intern. Emerg. Med. 2021, 16, 1, 201-207.
-
Z. Wang, A.B. Roberts, J.A. Buffa, B.S. Levison, W. Zhu, E. Org, X. Gu, Y. Huang, M. Zamanian-Daryoush, M.K. Culley, A.J. DiDonato, X. Fu, J.E. Hazen, D. Krajcik, J.A. DiDonato, A.J. Lusis, S.L. Hazen. "Non-lethal inhibition of gut microbial trimethylamine production for the treatment of atherosclerosis." Cell 2015, 163, 1, 1585-1595.
-
C.-Y. Chen, H.B. Leu, S.C. Wang, S.H. Tsai, R.H. Chou, Y.W. Lu, Y.L. Tsai, C.S. Kuo, P.H. Huang, J.W. Chen, S.J. Lin. "Inhibition of Trimethylamine N-oxide Attenuates Neointimal Formation Through Reduction of Inflammasome and Oxidative Stress in a Mouse Model of Carotid Artery Ligation." Antioxid. Redox Signal. 2023, 1-3, 38, 215-233.
-
H. Lee, X. Liu, J.-P. An, Y. Wang. "Identification of Polymethoxyflavones (PMFs) from Orange Peel and Their Inhibitory Effects on the Formation of Trimethylamine (TMA) and Trimethylamine-N-oxide (TMAO) Using cntA/B and cutC/D Enzymes and Molecular Docking." J. Agric. Food Chem. 2023, 71, 43, 16114-16124.
-
Z.L. Yu, L.Y. Zhang, X.M. Jiang, C.H. Xue, N. Chi, T.T. Zhang, Y.M. Wang. "Effects of dietary choline, betaine, and L-carnitine on the generation of trimethylamine-N-oxide in healthy mice." J. Food Sci. 2020, 85, 7, 2207-2215.
-
K. Fadhlaoui, M.-E. Arnal, M. Martineau, P. Camponova, B. Ollivier, P.W. O'Toole, J.-F. Brugère. "Archaea, specific genetic traits, and development of improved bacterial live biotherapeutic products: another face of next-generation probiotics." Appl. Microbiol. Biotechnol. 2020, 104, 11, 4705-4716.
-
R. El Hage, N. Al-Arawe, I. Hinterseher. "The Role of the Gut Microbiome and Trimethylamine Oxide in Atherosclerosis and Age-Related Disease." Int. J. Mol. Sci. 2023, 24, 3, 2399.
-
M. Dwidar, J.A. Buffa, Z. Wang, A. Santos, A.N. Tittle, X. Fu, A.M. Hajjar, J.A. DiDonato, S.L. Hazen. "Assembling the anaerobic γ-butyrobetaine to TMA metabolic pathway in Escherichia fergusonii and confirming its role in TMA production from dietary L-carnitine in murine models." mBio 2023, 14, 5, e00937-23.
-
J. Le Bris, N. Chen, A. Supandy, O. Rendueles, D. Van Tyne. "Phage therapy for Klebsiella pneumoniae: Understanding bacteria-phage interactions for therapeutic innovations." PLoS Pathog. 2025, 21, 4, e1012971.
-
T.W. Benson, K.A. Conrad, X.S. Li, Z. Wang, R.N. Helsley, R.C. Schugar, T.M. Coughlin, C. Wadding-Lee, S. Fleifil, H.M. Russell, T. Stone, M. Brooks, J.A. Buffa, K. Mani, M. Björck, A. Wanhainen, N. Sangwan, S. Biddinger, R. Bhandari, A. Ademoya, C. Pascual, W.H. Tang, M. Tranter, S.J. Cameron, J.M. Brown, S.L. Hazen, A.P. Owens III. "Gut microbiota-derived trimethylamine N-oxide contributes to abdominal aortic aneurysm through inflammatory and apoptotic mechanisms." Circulation 2023, 147, 14, 1079-1096.
-
Y.E. Gencay, D. Jasinskyte, C. Robert, S. Semsey, V. Martínez, A.Ø. Petersen, K. Brunner, A. de Santiago Torio, A. Salazar, I.C. Turcu, M.K. Eriksen, L. Koval, A. Takos, R. Pascal, T.S. Schou, L. Bayer, T. Bryde, K.C. Johansen, E.G. Bak, F. Smrekar, T.B. Doyle, M.J. Satlin, A. Gram, J. Carvalho, L. Jessen, B. Hallström, J. Hink, B. Damholt, A. Troy, M. Grove, J. Clube, C. Grøndahl, J.K. Haaber, E. van der Helm, M. Zdravkovic, M.O.A. Sommer. "Engineered phage with antibacterial CRISPR-Cas selectively reduce E. coli burden in mice." Nat. Biotechnol. 2024, 42, 2, 265-274.